This weekend, I have participated in the hack from home hackathon against COVID-19. I have joined a team led by Meabh MacMahon, a postdoctoral scientist at The Milner Institute, specialized in drug reuse. The idea that originated from Namshik Han was to use the knowledge gathered so far on drugs that are suspected to be efficient against COVID-19 to find other candidates that could also be efficient. I acted as an expert in machine learning in this hackathon. The team was composed of:
- Meabh MacMahon
- Namshik Han
- WooChang Hwang
- Justin Barton
- Mukunthan Tharmakulasingam
- Paul Bilokon
- Priya Gaddi
- Soorin Yim
- and me, of course.
The wonderful world of drugs and chemistry
I had no idea how drug research worked before the hackathon. I read some literature and discovered that several other actors were on the same subject and had already identified quite a lot of candidates. Most of them focus on finding drugs that can bind with Angiotensin-converting enzyme 2 — or ACE 2 — one of the main entry points of coronaviruses. Doing better than specialists in the field in a 2-day hackathon? Why not! It was a bit like playing the lottery.
For people interested in already existing work, you can browse:
- Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. — Liu, C. et al. (2020).
- Repurposing therapeutics for COVID-19: Supercomputer-based docking to the SARS-CoV-2 viral spike protein and viral spike protein-human ACE2 interface. — Smith, M., & Smith, J. C. (2020).
- A SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug-Repurposing. — Gordon, D. E., et al. (2020).
So what can an engineer that knows nothing about all of that do? As usual, do the boring stuff nobody wants to do and help! First, I had to get familiar with the sources of data. I was told that the reference database for drugs was ChemBL. It contains data about the drugs, their composition, what they are used, and all the associated clinical trials. As usual, I discovered that the data on the website was sometimes not filled, that there were duplicates, and actually that crawling a galaxy of websites was necessary to get a broad panorama of all the data available:
- ChemBL is focused on drugs and provides information about the associated trials
- PubChem is a broader database that contains chemical structure and related information for a wide range of chemical compounds, not only drugs
- Uniprot is the reference website for all proteins structures Since the hackathon is short, I mainly focused on ChemBL.
In term of software, I have used:
- the python client for ChemBL
- Indigo, a molecule manipulation software that I have first used to compute similarities between compounds
- RDKit, a second software that provides similarity computation, among other things
Finding candidates similar to COVID-19 fighters
Our plan for this hackathon was simple:
- Make a list of all drugs proven as efficient against COVID-19, in-vitro or in-vivo
- Compute the structural similarity between these compounds and all the other drugs available on the market
- Once this basic approach is made, try to refine the notion of similarity:
- Identify the targets of each drug, which site they are binding to
- Expand our research to drugs not already available on the market, maybe veterinary drugs
- Use existing embeddings to compute similarity
- Frame this problem as a machine learning one, and try to infer which drug can be useful against COVID-19
After a review of the ongoing studies, we have identified the following drugs as potentially efficient: Amoxicillin, Anakinra, ASC09, Ascorbic Acid, Azithromycin, Azvudine, Baloxavir, Baricitinib, Bromhexine Hydrochloride, Camostat Mesilate, Ceftriaxone, Chloroquine, Chloroquine diphosphate, clavulanate, cobicistat, Colchicine, Danoprevir, Darunavir, Deferoxamine, Dexamethasone, Dihydroartemisinin, Favipiravir, Fingolimod, Glucose, Hydrocortisone, Hydroxychloroquine, Hydroxychloroquine Sulfate, IFX-1, Leflunomide, Levofloxacin, Lopinavir, Losartan, Methylprednisolone, N-acetylcysteine, Nitric Oxide, Nitrogen, oseltamivir, Piclidenoson, Piperacillin, piperaquine, Plaquenil, Remdesivir, ribavirin, ritonavir, Ruxolitinib, Suramin, tazobactam, Tetrandrine, Thalidomide, Thiazide, thymosin alpha 1, TMC-310911, Tofacitinib, Triazavirin, Umifenovir, Vitamin C.
In drug processing, the structural similarity of molecules is computed by taking a fingerprint of the molecules and then calculating the Tanimoto coefficient between them. There are several ways to compute the fingerprint depending on the usage — similarity, subsequence matching, etc. Here is a small paper motivating these choices: Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations?. — Bajusz, D.,et al. (2015). — Journal of cheminformatics, 7(1), 20.
Results using similarity fingerprinting and Tanimoto coefficient
I use the similarity fingerprinting and similarity computation from the indigo package.
I first tried to plot this using a chord diagram. Unfortunately, there is no good python package to do that. I have used the bokeh function to do it, but I am not very satisfied. Here is a sample of the data. Uppercase compounds are conventional drugs, lowercase ones are our COVID-19 candidates.
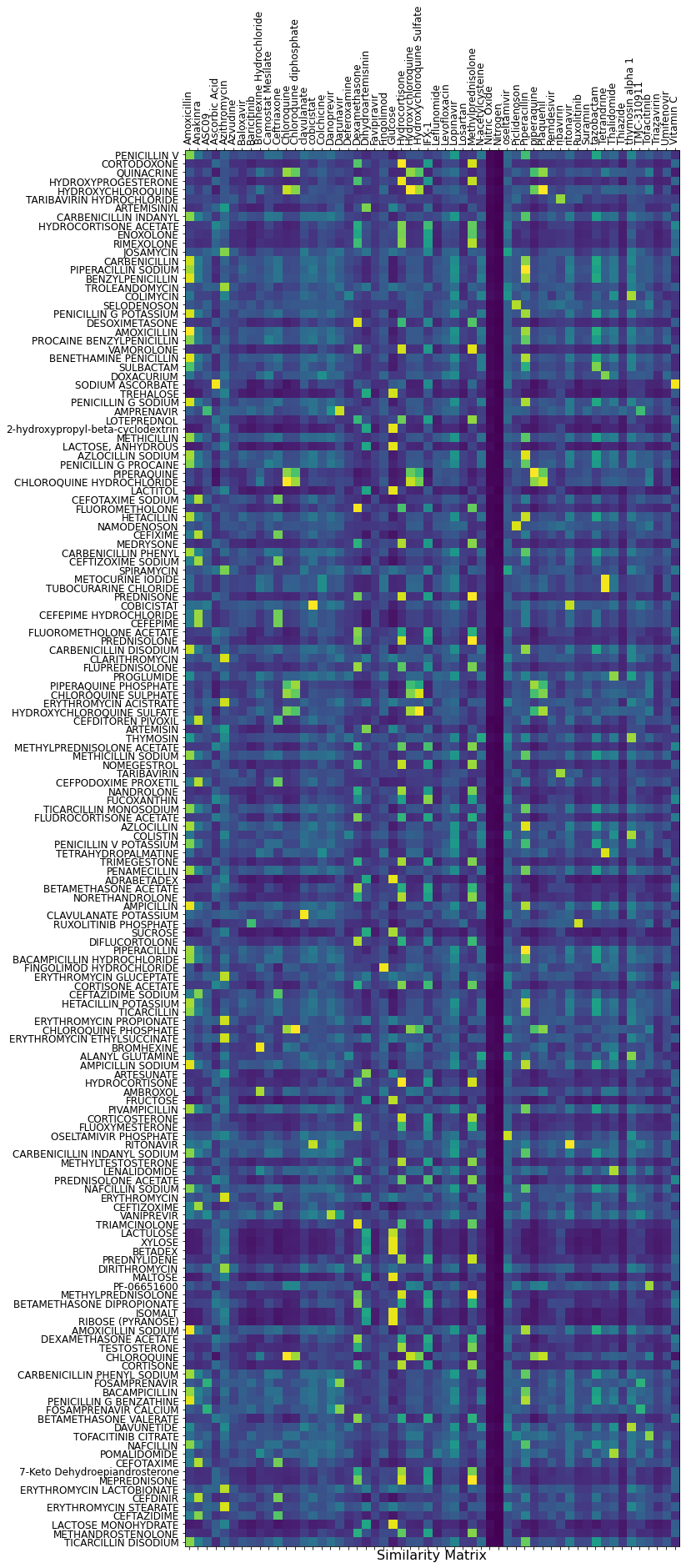
The second option is to display the full similarity matrix! It is clearly more comfortable to read. The column compounds are the COVID-19 candidates.
And finally, there is nothing like a plain old text output. Here are the compounds similar to Dexamethasone:
Dexamethasone
FLUOROMETHOLONE(0.982)
TRIAMCINOLONE(0.965)
DESOXIMETASONE(0.945)
DIFLUCORTOLONE(0.881)
FLUOXYMESTERONE(0.857)
FLUPREDNISOLONE(0.847)
DEXAMETHASONE ACETATE(0.846)
BETAMETHASONE ACETATE(0.846)
BETAMETHASONE VALERATE(0.821)
BETAMETHASONE DIPROPIONATE(0.821)
FLUOROMETHOLONE ACETATE(0.818)
FLUDROCORTISONE ACETATE(0.803)
Some of these compounds have already been tested, but surprisingly we have found no record of tests using Triamcinolone. This one seemed interesting enough to try to check it.
Results using Smiles2Vec and cosine similarity
We have used precomputed embeddings coming from this repository. It is basically a Word2Vec trained considering the molecules as sentences. For reference: [A novel methodology on distributed representations of proteins using their interacting ligands.] — Öztürk, H., et al. (2018). — Bioinformatics, 34(13), i295-i303.
The results do not seem that good. In particular when we look at this compound:
Amoxicillin
URIDINE TRIPHOSPHATE(0.420)
TIVOZANIB HYDROCHLORIDE(0.419)
PENTAERYTHRITOL TETRANITRATE(0.407)
INCB-9471(0.407)
TACRINE(0.404)
MELITRACEN(0.403)
TARLOXOTINIB BROMIDE(0.401)
We would expect to see Ampicillin as both molecules are close both in structure and function.
Partial conclusion
I have tried to vary different parameters like the distance, the similarity function, but it seems that we need to add some info to move forward. Most of the other studies use protein structure, but I cannot afford that since I do not have neither the data nor the expertise or the computational power.
In the end, we have selected Triamcinolone, Moxidectin, and Spinosad. These should be tested in-vitro in the coming week.
Work in progress: Using target data
ChemBL indicates the medical indication for all drugs and also more precisely, the proteins that are targeted. I am currently trying to mix this information with the structural similarity to have better predictions, but so far, there is no significant improvement!
Stay tuned for the follow-up!